Case study · ATAC-seq
How does androgen (DHT) stimulation reshape the open-chromatin landscape of prostate cancer cells, and can the driving transcription factor be identified from accessibility alone?
Two questions. First, how a single dose of androgen (dihydrotestosterone) remodels the open-chromatin landscape of an androgen-driven prostate cancer line. Second, whether the transcription factor responsible for that remodelling can be read straight off the ATAC-seq data, without needing a matched RNA-seq experiment.
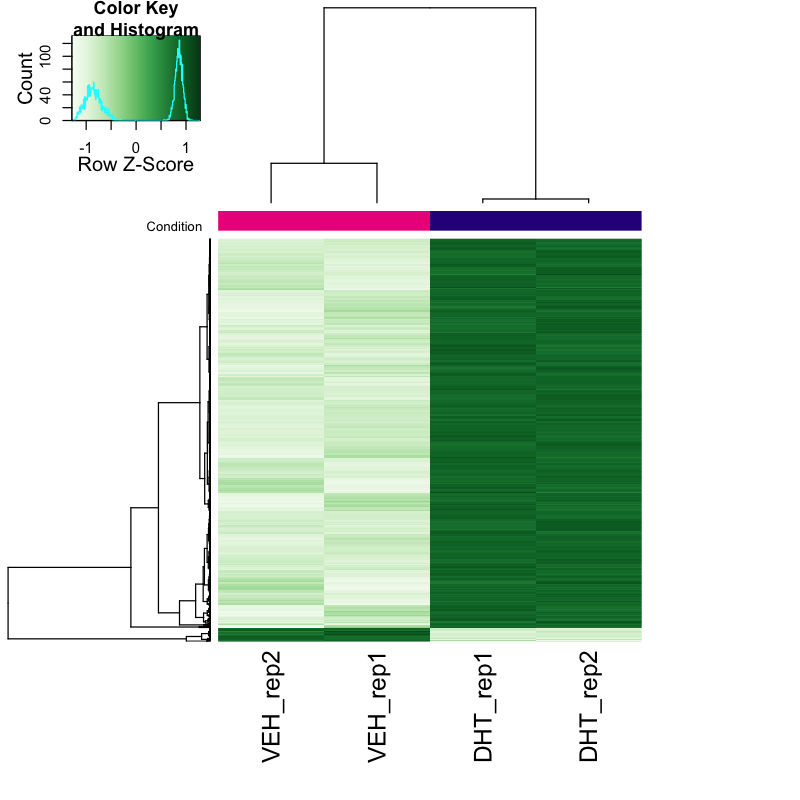
End-to-end ATAC-seq workflow on 4 samples (VCaP, vehicle vs 1 nM DHT, 2 replicates each): adapter trimming, Bowtie2 alignment, mitochondrial and ENCODE-blacklist filtering, MACS3 peak calling, and ENCODE-standard quality control (TSS enrichment, fragment-size periodicity, FRiP, library complexity). A fixed-width reproducible consensus peak set then fed three layers of analysis: differential accessibility with DiffBind (DESeq2), per-transcription-factor motif activity with chromVAR, and base-resolution footprinting with TOBIAS, all against the JASPAR2024 motif set. Differentially accessible peaks were annotated to their nearest gene.
This is the part that is not a pipeline output. The numbers say chromatin opens far more than it closes and that AR is the most variable motif; the read is what that means. A hormone that opens thousands of enhancers in a single dose is switching on a pre-wired regulatory programme, not building one from scratch, which is why the pioneer factors FOXA1 and HOXB13 surface alongside AR: they mark the enhancers AR then occupies. That the answer falls straight out of accessibility, with no matched RNA-seq, matters biologically, because it locates the change at the level of regulation rather than of steady-state transcription. The report also states the honest limit: AR shares its response element with the glucocorticoid and progesterone receptors, so the motif evidence names the family, and it is the biological context (VCaP, DHT) that pins it to AR specifically. A generic tool would have handed back the same motif table and left that distinction unmade.
Interpretation ends by pointing at the next experiment, not by closing the file. Here the natural follow-ups are: a matched RNA-seq or nascent-transcription assay to confirm the newly opened enhancers actually drive their nearest genes; an AR ChIP-seq to separate direct AR binding from co-factor-driven opening; and an anti-androgen (e.g. enzalutamide) arm to test whether the same enhancer set closes on AR blockade, which is the therapeutically relevant question in prostate cancer. These are written into the report as future perspectives, so the deliverable is a starting point for the next round of work, not just a description of this one.
ATAC-seq is often delivered as a peak list and little else. The value here is the interpretation: three independent views of the same data converging on one answer, the androgen receptor programme, written into the open chromatin and named without any expression data, then placed in its biological context and pointed at the next experiment. The report states that conclusion in plain English, flags the one honest caveat, and keeps every number traceable to an auditable pipeline. This is the kind of regulatory-biology question, which transcription factor is driving the change, that ATAC-seq answers better than expression alone, and it is the read on top, not the peak calling, that a client is actually paying for.
The actual client deliverable layout — same files, same structure, same format.
Fixed-fee, 7–10 business days. Email with your omics type + sample count and a quote comes back within 2 business days.